
A team of researchers has unveiled a promising alternative to the conventional means of synthesizing ammonia, one that is more environmentally friendly.
Details of their research were published in the Journal of Materials Chemistry A on February 21, 2024.
When Fritz Haber and Carl Bosch invented a means to synthesize ammonia from nitrogen and hydrogen gas in the early 20th century, it enabled the production of the chemical at an industrial level. To this day, the Haber-Bosch synthesis remains the dominant means of producing ammonia.
Yet the method has some environmental drawbacks. It is energy and resource intensive, and producing hydrogen gas often involves natural gas, which releases carbon dioxide as a by-product.
The electrochemical nitrogen reduction reaction (ENRR), where nitrogen gas from the air can be converted into ammonia using an electrical current, is seen as a promising and sustainable alternative. Pursuing high-performance and cost-effective ENRR catalysts, however, is an open challenge for achieving commercial-scale ambient ammonia production.
"We explored the potential of less-precious transition metal disulfides (TMS2) as catalysts for ENRR," says Hao Li, associate professor at Tohoku University's Advanced Institute for Materials 雷速体育_中国足彩网¥在线直播 (WPI-AIMR) and corresponding author of the paper. "Through meticulous analysis of electrochemistry-induced surface states, we uncovered a previously unrecognized factor contributing to their high ENRR performance: S-vacancy generation."
Li and his colleagues started with a typical ENRR TMS2 catalyst, iron disulfide (FeS2), where they observed that under ENRR conditions, S-vacancies can be easily generated on the catalyst surface. Through advanced computational simulations, they demonstrated that this electrochemistry-driven "in situ" generation of S-vacancies significantly enhances ENRR activity by promoting stronger N-N adsorption and activation.
Experimental observations confirmed their findings, which were also consistent with recent literature on ENRR potential windows reaching maximum Faradaic efficiency - the measure of the effectiveness of an electrochemical process in converting electrical energy into chemical energy or vice versa.
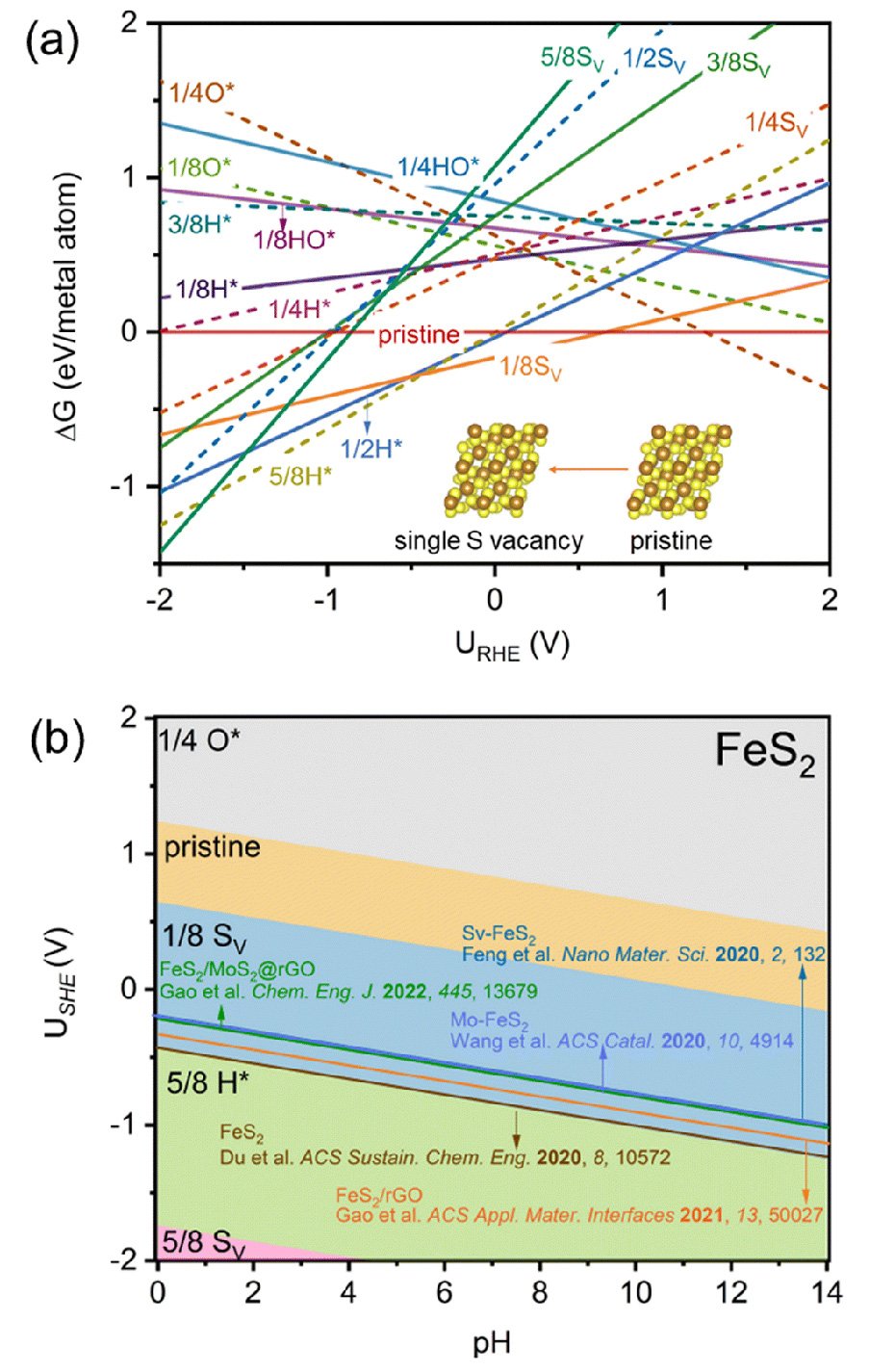
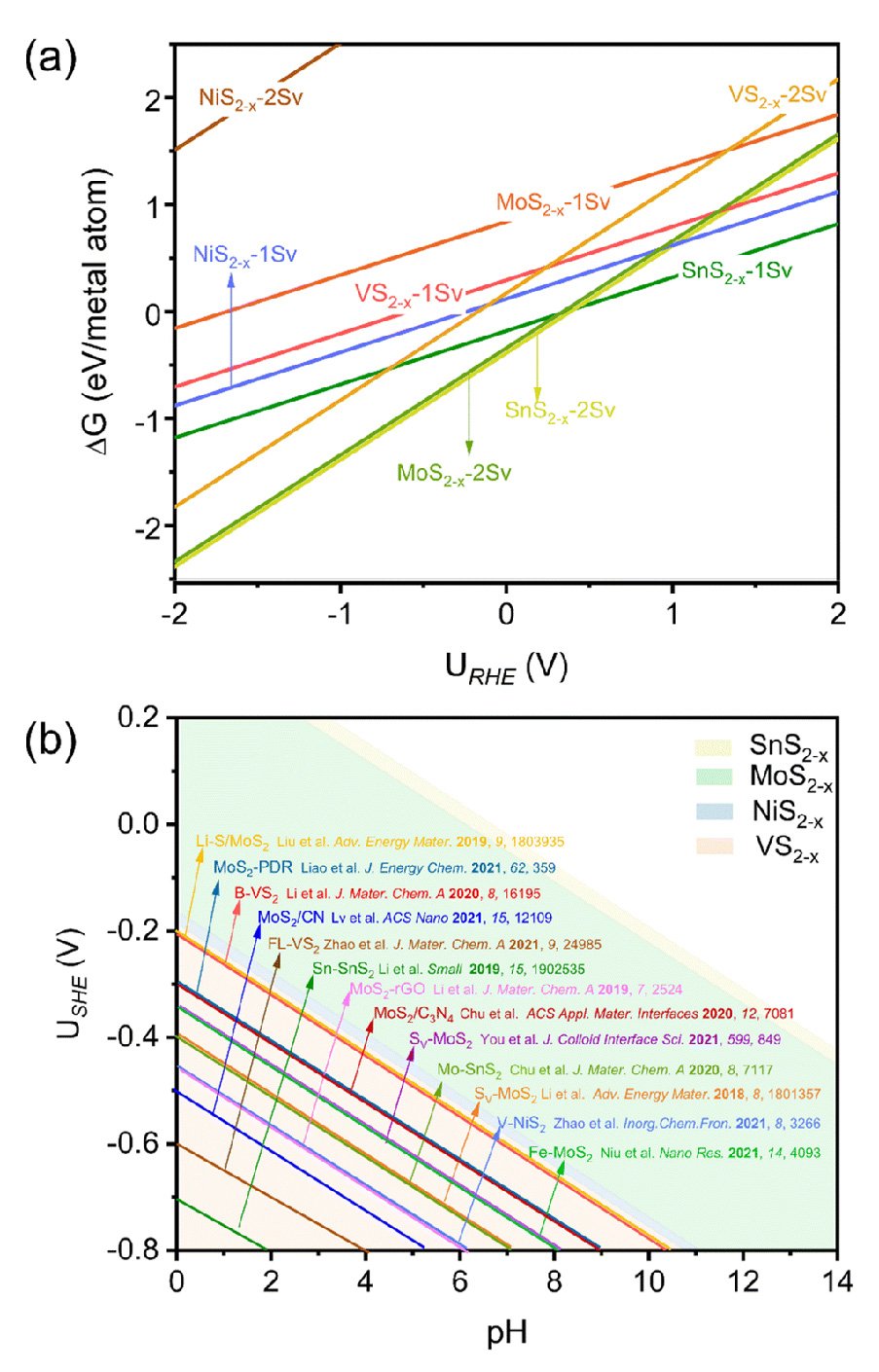
Their analysis also extended to other TMS2 catalysts (SnS2, MoS2, NiS2, and VS2), revealing a universal phenomenon of "in situ'' S-vacancy generation under ENRR potentials.
"Our research underscores the critical importance of considering surface states in the design of ENRR catalysts," adds Li. "By shedding light on the role of S-vacancies, we have provided a valuable roadmap for enhancing ENRR performance and accelerating the transition towards sustainable ammonia production."
This work was supported by AIMR Fusion 雷速体育_中国足彩网¥在线直播 and also received substantial support for the JSPS Postdoctoral Fellowship for Dr. Tianyi Wang in the Hao Li Lab.
- Publication Details:
Title: Origin of electrocatalytic nitrogen reduction activity over transition metal disulfides: critical role of in situ generation of S vacancy
Authors: Tianyi Wang, Zhongyuan Guo, Hirofumi Oka, Akichika Kumatani, Chuangwei Liu, and Hao Li
Journal: Journal of Materials Chemistry A
DOI: 10.1039/D4TA00307A
雷速体育_中国足彩网¥在线直播:
Hao Li,
Advanced Institute for Materials 雷速体育_中国足彩网¥在线直播 (WPI-AIMR), Tohoku University
Email: li.hao.b8 tohoku.ac.jp
tohoku.ac.jp
Website: https://www.li-lab-cat-design.com/